
African Journal of Respiratory Medicine received 855 citations as per google scholar report
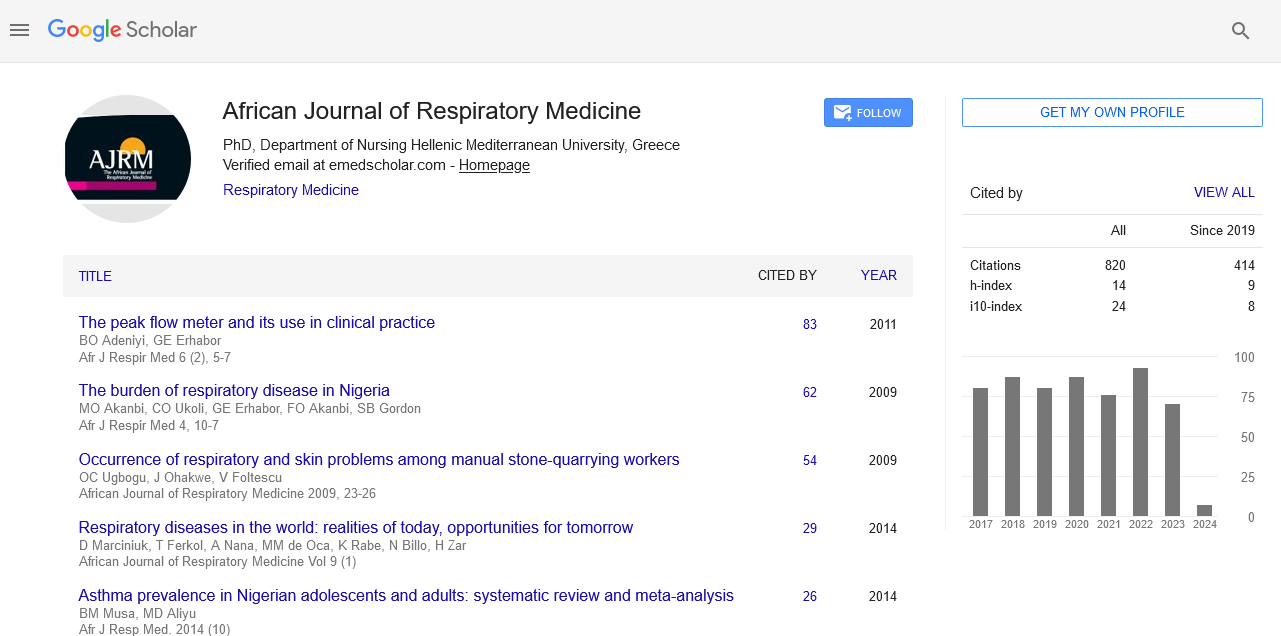
Research - (2022) Volume 17, Issue 4
Received: 28-Mar-2022, Manuscript No. ajrm-22-60058; Editor assigned: 30-Mar-2022, Pre QC No. ajrm-22-60058 (PQ); Reviewed: 13-Apr-2022, QC No. ajrm-22-60058; Revised: 18-Apr-2022, Manuscript No. ajrm-22-60058 (R); Published: 25-Apr-2022, DOI: 10.54931/1747-5597.22.17.16.
Background: Carcinoid tumors are a subset of neuroendocrine tumors, of which pulmonary carcinoid (PC) is the second most common type. PC is divided into typical (TC) and atypical (AC) subtypes. They have a low incidence and a relatively high survival rate. In this study, we report the characteristics, prognosis, and results of PC patients from Gansu Provincial People’s Hospital in China to better understand these tumors and their prognostic markers.
Materials and methods: This is a study based on retrospective chart review in our tertiary referral center. In this study, there were 36 consecutive PC patients enrolled from 2000 to 2017. The information collected including demographics, tumor characteristics and recurrence and survival rates. All patients had their last follow up visit on December 31, 2020.
Results: 39% of patients were asymptomatic at the time of diagnosis and, most patients suffer from early disease. 80% of patients have typical PC and 20% are atypical PC. Both subtypes have a good prognosis, but the atypical subtype has a higher rate of progression (60%). The median time to disease progression or recurrence is 6 years. Only 2 patients died. Immunohistochemistry (IHC) markers in those tested showed high sensitivity for chromogranin A (82%) and synaptophysin (100%).
Conclusion: Our study provides insights for PC patients with low incidence in Northwest China. Most patients are treated with surgery and have a good survival rate. IHC marking should be better implemented. Larger studies are needed to illustrate and verify our results.
Carcinoid; Pulmonary; Neuroendocrine tumors; Surgery
The Carcinoid tumors are a subset of neuroendocrine tumors. Most carcinoids originate in the gastrointestinal tract, and the prevalence is about 60%. The pulmonary system is the second most commonly affected system, with a prevalence of approximately 27%. Pulmonary neuroendocrine tumors represent a range, including the most aggressive small cell lung cancer (SCLC), lung carcinoid (PC), diffuse idiopathic pulmonary neuroendocrine cell hyperplasia (DIPNECH) and other less aggressive types Form. PC can secrete a variety of hormones, leading to carcinoid syndrome, variants of carcinoid syndrome, rare Cushing syndrome or endocrine syndrome produced by other peptides.1-4
Lung carcinoid tumors originated from Kulchitsky cells in the bronchopulmonary mucosa.5 According to its histological characteristics, PC is usually divided into two subtypes: classic lung carcinoid (TC) and atypical lung carcinoid (AC), with a prevalence of 9:1.1,2 TC shows high differentiation, rare mitosis (less than 3 mitosis per 10 high power fields), and rare pleomorphism or necrosis. However, AC increased cell atypia, moderate mitotic index (3 to 10 mitotic figures per 10 high power fields), and increased pleomorphism or necrosis.1 AC accounts for 10% of carcinoid tumors, mainly located in the periphery of the lung. AC is larger than TC, is more aggressive, and metastasizes more frequently.5-7
Although their incidence in the United States is very low, about 5.25 cases per 100,000 people, carcinoid tumors can cause deaths in 5% to 25% of patients.1,6 Compared with atypical carcinoids, typical carcinoids have a better overall survival rate.8 The role of chemotherapy and radiotherapy in PC is controversial, and the current prognostic significance of tumor staging and histological type is still limited.6
Diagnostic tests for PC currently include the presence of tumor markers. Chromogranin A and synaptophysin are glycoproteins used as neuroendocrine tumor markers in serum and tissues, respectively. They belong to the neuron specific enolase group and are considered to be evidence of the neuroendocrine origin of tumors. The immunostaining of chromogranin A antibody varies depending on the origin of the carcinoid tumor. For example, chromogranin A antibody cannot stain 40% of hindgut tumors and 12% of foregut carcinoid tumors.8 However, synaptophysin stains all carcinoid tumors, regardless of their origin, and is very specific and sensitive.
A key point in the proper management of pulmonary neuroendocrine tumors is a multidisciplinary approach. According to the individual patient and the disease site, for patients with advanced or metastatic disease, surgical treatment of primary tumors and oligometastatic PCs should be considered to achieve the purpose of curing or controlling symptoms. In addition, when considering systemic therapy, in some cases, low grade typical and atypical PCs have shown a certain response to multiple combination chemotherapy drugs in historical data, such as temozolomide with or without capecitabine, Cisplatin or carboplatin combined with etoposide.9,10It is usually recommended for progressive metastases without other treatment options. Another option for controlling PC symptoms and slowing the progression of the disease is somatostatin analogs, such as octreotide, lanreotide, or pasireotide, because low grade pulmonary neuroendocrine tumors often exhibit surface overexpression of somatostatin receptors. These analogs bind with high affinity to hinder the secretion of peptides and amines that cause symptoms and tumor growth.11-14 In addition; there are other options for systemic treatment of PC, including peptide receptor radioligand therapy (PRRT). PRRT is a process of delivering radiolabeled drugs (such as 90Y-labeled octreotide or 177Lu-DOTATATE) to specific targets (such as somatostatin receptors).15 In addition, given the increased activation of the mTOR signaling pathway in pulmonary neuroendocrine tumors, everolimus is a mTOR (mechanical target of rapamycin) inhibitor and a potential therapy approved for patients with advanced lung NET.16,17 Interferon alpha and anti-angiogenic drugs (such as bevacizumab) are also recommended for the treatment of patients with advanced advanced lung NET (such as PC).18-21
On the one hand, the slow process of PC disease, on the other hand, the scarcity of randomized clinical trials to study the best management and prognosis of PC, emphasize the need to better understand these tumors. In this study, we report the characteristics, prognosis, and results of PC patients from Gansu Provincial People’s Hospital in China to better understand these tumors and their prognostic markers.
We conducted a retrospective search of all medical records to identify lung carcinoid patients diagnosed and treated at Gansu Provincial People’s Hospital from 2000 to 2017. The patient has agreed that his doctor has the right to contact for follow up in the future up. We collected information about demographics, tumor characteristics, staging, and recurrence from medical charts. All patients were treated as the last follow up survival status of this study on December 31, 2020. All statistical analyses were performed using SPSS Statistics version 20.0.
Patient characteristics
A total of 36 patients were enrolled in this study, of which 53% were women. The median age at diagnosis is 50 years (19-71). 13 patients (36.1%) were smokers. Two patients (5.6%) had asthma or allergic airway disease, and two patients (5.6%) had a family history of malignant tumors.
Disease characteristics and clinical aspects
Fourteen patients (39%) were asymptomatic at the time of diagnosis. 22 patients (61%) had symptoms such as coughing, dyspnea, and hemoptysis. 23 patients (63.89%) had lung carcinoids involving the right lung. The lesion data of 34 patients were retrieved, of which 29 patients (85.3%) had a single lesion. The largest tumor diameter recorded was 2.6 cm. 28 patients had tumors with typical subtypes, and 7 had atypical subtypes. 87% of patients have local disease, and 13% of patients have metastatic disease. Pathological evaluation of surgical specimens showed that most patients (84.2%) had no lymphatic vascular invasion (LVI), and 22.7% of patients had positive lymph node involvement (Table 1).
| Characteristics | N | % |
|---|---|---|
| Smoking Status | ||
| Smokers | 13 | 36.1 |
| Non-smoker | 23 | 63.9 |
| Gender | ||
| Male | 17 | 47.2 |
| Female | 19 | 52.7 |
| Focality | ||
| Unifocal | 29 | 85.3 |
| Multifocal | 5 | 14.7 |
| Histologic type | ||
| Typical | 28 | 80 |
| Atypical | 7 | 20 |
| Lymphovascular invasion | ||
| Absent | 16 | 84.2 |
| Present | 3 | 15.8 |
| Status on last follow up | ||
| Stable | 29 | 80.5 |
| Progression | 2 | 5.6 |
| Recurrence | 3 | 8.3 |
| Deceased | 2 | 5.6 |
In our patient population, only 17 patients were tested for chromogranin A, and 14 patients (82.35%) were positive.
Only 16 patients were tested for synaptophysin status, and all patients were positive (Figure 1).
Figure 1. Biomarker test for chromogranin A and Synaptophysin. Chromogranin A is 82.35% and Synaptophysin is 100% positive in PC patients.
Disease management
Patients are diagnosed by CT-guided biopsy or endobronchial ultrasound guided biopsy. The staging of the attending physician is based on the American Joint Committee on Cancer (AJCC) staging system. In addition to patients with stage IV disease, 27 patients (75%) underwent surgical resection. Only 5 patients (14%) received chemotherapy in a metastatic setting. One patient received radiation therapy in adjuvant therapy. Different regimens include cisplatin and etoposide, docetaxel, gemcitabine, and capecitabine.
Patient follow up
The median follow up time was 6.17 years. The median overall survival is 6 years. The average time to progress or relapse was 9.67 years, and the median time to progress or relapse was 6.17 years. The last follow up time was December 31, 2020, and 34 patients (94.5%) were alive. 29 patients (80.5%) had disease free or stable disease, 2 patients (5.5%) had disease progression, and 3 patients (8.5%) had disease recurrence. Finally, in 2 patients (5.5%) who died, the death was attributed to disease progression.
Of the 28 patients with typical carcinoid tumors, 25 (89.28%) had no disease progression or recurrence. Among the remaining patients, 1 patient developed disease progression after 43 months and had stage IV disease since diagnosis. The second patient progressed 55 months later and died in progress. The third patient progressed 80 months later and was diagnosed with stage IA2 disease.
Among the 7 patients with atypical carcinoids, 4 patients (57%) had disease progression or recurrence. One patient was in stage IV at the time of diagnosis and progressed and died 7 months later. Another patient progressed after 43 months and was diagnosed as stage II B. In addition, the third patient was in stage III A at the time of diagnosis and progressed 74 months later, while the fourth patient was in stage I A1 at the time of diagnosis and progressed after 17 years (Tables 2 and 3).
| Histologic Type | Progression or Recurrence | ||
|---|---|---|---|
| Yes | No | ||
| Typical | 28 | 3 | 25 |
| Atypical | 7 | 4 | 3 |
| Not Available | 1 | 0 | 1 |
| Total | 36 | 7 | 29 |
| Histologic Type | Deceased on follow-up | ||
|---|---|---|---|
| Yes | No | ||
| Typical | 28 | 1 | 27 |
| Atypical | 7 | 1 | 6 |
| Not Available | 1 | 0 | 1 |
| Total | 36 | 2 | 34 |
Lung carcinoid is a rare entity of neuroendocrine tumors, including many histopathological variations in tumor biology and prognosis. Age at diagnosis, histopathological type, and the presence of mediastinal or subcarinal lymph nodes represent independent prognostic factors for overall survival, relative survival, and event free survival of lung carcinoids.8 Our study was conducted for the first time in Northwest China. It reviewed the charts of 36 PC patients and evaluated their clinical characteristics, clinical progress, and survival rate at a median follow up of 6 years.
In our study, we found that the 5 year survival rate of lung carcinoid patients in Northwest China is 94.5%, which is comparable to the reported US studies of 88% and 96% of 102. PC patients studied by Daskalis et al. in Europe.22,23
Compared with patients with typical carcinoids, patients with atypical carcinoids usually have a more aggressive course, a worse prognosis, and a higher rate of lymph node metastasis and recurrence.1 A study by Song et al.24 An analysis of 68 AC patients showed that the 5 year and 10 year overall survival rates were 70.6% and 61.8%, respectively. Another study by Chong et al.3 Reported even lower 5 year and 10 year overall survival rates of 35% and 44%, respectively. There are 7 AC patients in our study, and only one patient died a few months after diagnosis. This makes the 5 year overall survival rate of our AC population 86%, which is higher than the published data. On the other hand, TC generally has a good prognosis, with 5 year and 10 year survival rates of 90% and 84%, respectively. Our patient population shows that the 5 year overall survival rate of TC is 96.5%, which is also better than reported in the literature. Perhaps our higher survival rate among patients with AC and TC is due to the high proportion of young people (age range 19-46 years), health status, and ultimately the early diagnosis stage of most patients in our population, which has been better than before. The survival rate. However, regarding the progression rate, our results are comparable to published data. The recurrence rate of TC is 11% (3 out of 28 patients), while the recurrence rate of AC is 57% (4 out of 7 patients).8,23 All patients who progressed are non-smokers, and none of the patients in the early stages were affected by LVI or LN, which may mean that these factors have nothing to do with the prognosis of PC. Nevertheless, patients with an earlier stage of disease at the time of diagnosis do require a longer duration to reach up to 17 years, which suggests that it is associated with recurrence.
In addition, the high positive rates of chromogranin A and synaptophysin tested in our patients are similar to published data, showing high diagnostic sensitivity of 82% and 100%, respectively. Unfortunately, not all patients have been tested, which may reflect the limited use of these markers in current practice when diagnosing PC in our region. Another reasonable explanation may be to postpone the clinical use of these markers until the new millennium. Other markers with proven prognostic value, such as cyclin A2 and B1 of Brcic et al. or Aly et al. Spread through airspace (STAS). Or the prognostic value of little significance, such as Ki67, was not used in our study population.25-27
Most of our patients received surgical resection (n=27, 75%); 14% received chemotherapy and 3% received radiotherapy. Surgical resection is still the gold standard treatment for PC, and the vast majority of patients in the case series remain relapse free for many years after surgery. However, there are controversies regarding the optimal scope of surgery and the prognostic significance of systemic chemotherapy and radiotherapy.3 In our patient population, 13 patients (36.1%) were smokers and most of them had typical histopathology. However, it has been reported that AC rather than TC is usually associated with tobacco use.1,4 Two patients (5.6%) had asthma or allergic airway disease, 39% were asymptomatic at the time of diagnosis, and 61% had respiratory symptoms, including cough and pneumonia. These findings were compared with published data on clinical manifestations including cough, recurrent pneumonia, dyspnea, hemoptysis, wheezing, and fever. In contrast, the diagnosis of some patients may be entirely accidental and show no symptoms.
Our research has some limitations. The first limitation is the retrospective design of the research. Since the information relies on chart review, incomplete documentation and data retrieval have been introduced. When analyzing specific variables (including histological type, lymphatic vascular invasion, and management), the small sample size is another limitation. However, our research has two important advantages. First, considering that the incidence of the disease in the United States is as low as 5,25 cases per 100,000 cases, the sample size is relatively sufficient. 1,6 Secondly, as far as we know, this study is the first in Northwest China, and there are few data on the prevalence and management of lung carcinoids in this area.28-31
Lung carcinoids (except SCLC) are rare neuroendocrine tumors with a good overall prognosis. Their research in our area is insufficient. In our study population, the 6 year median overall survival rate for typical and atypical lung carcinoids is very good, with 5 year overall survival rates of 96.5% and 86%, respectively. TC is associated with a lower recurrence rate (11% for TC and 57% for AC). This means that the carcinoid patient population in Northwest China is similar to the published population in the world in terms of survival, recurrence and diagnosis. Smoking, LVI and LN status do not affect the results, but early diagnosis may play a role in the prognosis. Chromogranin A and synaptophysin are good diagnostic markers and should be used more frequently, other prognostic markers including cyclin and STAS were not used, and it is impossible to draw conclusions about their prognostic impact on the population of Northwest China from our study.
In conclusion, more extensive research is needed to better understand the prevalence of PC, the importance and role of different markers in PC, and the best management of such diseases in Northwest China. Perhaps the establishment of a joint collaborative research network between referral centers in our region can help conduct more research and include more patients with this rare disease, and provide funding for the use of more prognostic markers.
This work was supported by the Youth Project of Science Research Fund of People’s Hospital of Gansu Provincial (No.18GSSY5-17).
LPH and YJS designed the work, LPH, ZB, LAL and CD wrote the manuscript. All authors read and approved the final manuscript.
All the authors agree to publish this article.
The authors declare that they have no competing interests.
Select your language of interest to view the total content in your interested language
To read the issue click on a cover

